Acțiune:



Sindromul Smith-Lemli-Opitz Simptomele, cauzele și tratamentul
Sindromul Smith-Lemli-Opitz (SLO) este o tulburare metabolică, care cuprinde mai multe simptome diferite, inclusiv o creștere semnificativă lentă, trăsăturile caracteristice ușor, microcefalie (cap de măsurare mai mică decât normal), retard mental care poate fi dificultăți ușoare sau moderate de învățare și probleme comportament.
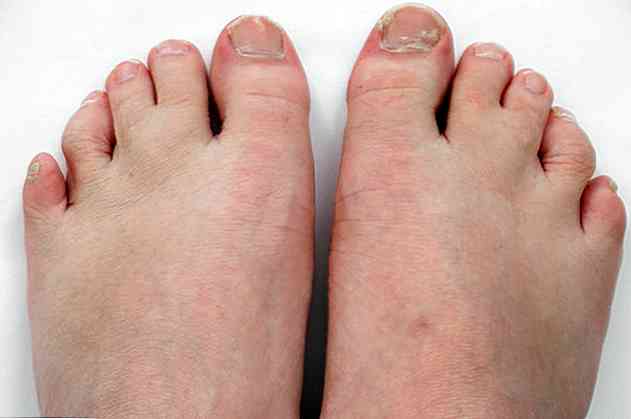
Este, de asemenea, însoțită de malformații în plămâni, inimă, rinichi, intestin și chiar în organele genitale. În plus, ele pot prezenta sindectilie sau fuziune a unora dintre degete sau polidactilă; ceea ce înseamnă că au mai mult de 5 degete pe un picior sau pe o mână.
Se pare că se datorează lipsei unei enzime care este importantă pentru metabolizarea colesterolului care este dobândită prin moștenirea genetică a modelului autosomal recesiv.
Cu toate acestea, astfel de prezentări par a varia foarte mult în funcție de severitatea bolii chiar și în aceeași familie.
Acest sindrom poate apărea în literatura de specialitate cu nume cum ar fi deficiența de 7-dehidrocolesterol reductază, sindromul RSH sau sindromul SLO.
O mică istorie ...
În 1964, pediatri David Smith, Luc Lemli și John Opitz a descris un 3 pacienți de sex masculin cu microcefalie și hypogenitalism, și această condiție este definită ca RSH prin inițialele numelor originale ale acestor pacienți.
Ulterior, numele sindromului a fost modificat la numele de familie al descoperitorilor (SLO).
Aproximativ 30 de ani mai târziu, Tint et al. (1994) a descoperit la 5 pacienți cu această afecțiune concentrații semnificativ scăzute ale colesterolului din sânge, dar o creștere de peste 1000 de ori a nivelurilor de 7-dehidrocolesterol. Ei au văzut că această creștere sa datorat lipsei unei enzime care ar trebui să transforme 7-dehidrocolesterolul în colesterol.
Ulterior, gena DHCR7 asociată cu această boală a fost identificată, clonând în 1998 (Witsch-Baumgartner & Lanthaler, 2015).
statistică
Sindromul Smith-Lemli-Opitz afectează aproximativ 1 din 20.000 până la 60.000 de născuți vii din întreaga lume. Puteți obține într-adevăr pentru a rula în 1 în 1590 la 13500 de oameni, dar această cifră nu este folosit, deoarece mulți fetuși cu aceasta conditie mor inainte de nastere (Organizația Națională pentru bolile rare, 2016).
În ceea ce privește sexul, afectează atât bărbații, cât și femeile în mod egal, deși este mai ușor de diagnosticat la bărbați, deoarece malformațiile genitale sunt mai vizibile decât la femei. În plus, se pare că are o frecvență mai mare în rândul persoanelor de origine europeană; în special din țările aparținând Europei Centrale, cum ar fi Republica Cehă sau Slovacia. Cu toate acestea, este foarte rar în populația din Africa sau Asia.
Cauzele sindromului Smith-Lemli-Opitz
Sindromul SLO apare datorită mutațiilor din gena DHCR7, prezentă pe cromozomul 11, care este responsabilă pentru trimiterea ordinelor pentru fabricarea enzimei 7-dehidrocolesterol reductază. Aceasta este enzima care modulează producția de colesterol și ar fi absentă sau foarte mică în acest sindrom, ceea ce duce la o producție insuficientă de colesterol care ar împiedica creșterea normală.
Acest lucru produce un mare impact deoarece colesterolul este important în organism. Se compune dintr-o lipidă similară grăsimii, obținută în principal de alimente de origine animală, cum ar fi gălbenușurile de ou, produsele lactate, carnea, păsările de curte și peștii.
Este esențial ca embrionul să se dezvolte fără dificultate, având funcții importante, cum ar fi contribuția la structura membranelor celulelor și a mielinei (substanță care acoperă celulele creierului). De asemenea, servește la producerea de hormoni și acizi digestivi.
Lipsa enzimei 7-dehidrocholesterol reductazei determină acumularea în corp a componentelor care pot fi toxice pentru colesterol. Așadar, avem, pe de o parte, niveluri reduse de colesterol și, în același timp, acumularea de substanțe care pot fi toxice pentru organism; cauzând lipsa creșterii, întârzierea mintală, malformațiile fizice și problemele din organele interne.
Cu toate acestea, nu se știe cu certitudine cum aceste probleme asociate cu colesterolul dau naștere la simptomatologia sindromului Smith-Lemli-Opitz.
Au fost găsite acum în sindromul legate mai mult de 130 de gene DHCR7, într-adevăr mutații, este o bază de date în cazul în care toate cazurile descrise cu variante SLO, sunt incluse fenotipuri și genotipurile lor.
Deși există atât de multe mutații posibile, majoritatea cazurilor aparțin celor 5 cele mai frecvente, iar restul sunt foarte rare (Witsch-Baumgartner & Lanthaler, 2015).
Aceste mutații ale genei DHCR7 sunt mostenite intr-un model autosomal recesiv, ceea ce înseamnă că o persoană care să prezinte sindromul trebuie să fi moștenit gena mutant de la ambii părinți. Dacă îl primiți numai de la unul dintre părinți, nu veți prezenta boala; dar ar putea fi un transportator și să o transmită în viitor.
Există un risc de 25% ca cei doi transportatori ai părinților să aibă un copil afectat, în timp ce riscul ca copilul să fie transportator ar fi, de asemenea, de 50% în fiecare sarcină.Pe de altă parte, în 25% din cazuri se poate naște fără aceste mutații genetice sau poate fi un purtător; toate aceste date fiind independente de sexul copilului (Organizația Națională pentru Tulburări Rare, 2016).
Trebuie avut în vedere faptul că există o probabilitate mai mare de a avea copii cu tulburare genetica recesiva daca orice parinti care sunt rude apropiate (sau sânge) pentru părinții care nu au aceste link-uri.
Ce simptome are?
Simptomele variază în funcție de persoana afectată, în funcție de cantitatea de colesterol pe care o pot produce.
Potrivit lui Jiménez Ramírez et al. (2001), caracteristicile clinice acoperă mai multe aspecte și pot fi foarte diverse. În general, ele se găsesc pe față, membre și organe genitale; deși pot implica alte sisteme corporale.
Mulți dintre cei afectați au caracteristici tipice ale autismului, afectând interacțiunea socială. Dacă starea este ușoară, pot fi văzute doar câteva probleme legate de învățare și comportament; dar, în cazurile severe, persoana poate avea o mare dizabilitate intelectuală și anomalii fizice, care pot duce la deces.
Există simptome care pot fi deja prezente de la naștere a individului, dar vom include pe cele care au loc în toate etapele vieții:
La mai mult de 50% dintre pacienți:
- Lipsa dezvoltării fizice observată după naștere.
- Retardarea mentală (100%).
- Microcefalie (90%).
- Syndactyly sau fuzionarea a 2 sau 3 toe (<95%).
- Ptoza palpebrală, adică având unul din pleoapele superioare (70%).
- meat situat într-un loc diferit la bărbați normali, cum ar fi în partea de jos a conexiunii glandului sau trunchi intre scrot si penis. Acesta este prezent în 70% din cazuri.
- Cleștele palatului, care se manifestă ca un fel de gaură alungită în palat (50%).
- maxilarul redus sau micrognatia.
- Limba foarte mică (microglossia).
- Urechi de implantare scăzută.
- Nasul scurt.
- Coborârea incompletă a unuia sau a ambilor testicule.
- Hipotonie sau tonus muscular scăzut.
- Tulburări de alimentație.
- Tulburări comportamentale: comportamente antisociale, auto-distructive și violente. Autismul auto-stimulativ comportamente, de asemenea, apar ca mișcări repetitive de echilibrare.
- Autism
De la 10 la 50% din cazuri:
- Cataracta precoce.
- Polydactyly sau cu încă un deget după degetul mic.
- Întârzierea creșterii în faza fetală.
- Genitale ambigue.
- Defectele cardiace.
- rinichi multicystic.
- Absența unui rinichi sau ambele la naștere.
- boli hepatice.
- Hiperplazia suprarenală
- Anomalii pulmonare.
- Excesul de transpirație.
- anomalii cerebrale în structurile lor de la linia mediană și dezvoltarea incompletă a calos, sept și vermis cerebelos.
- acrocianoza: vasoconstricție cutanată care provoacă o culoare albăstruie în mâini și picioare.
- Picioarele lui Equinovar.
- stenoza pirocică (15%);
- boala Hirschprung, care cauzează o lipsă a motilității intestinale (15%);
- Fotosensibilitate.
Alte simptome:
- Obezitate sau comă.
- acumularea de lichid în corpul fătului.
-Alterații în dezvoltarea neurologică.
- Probleme neuropsihiatrice, care apar mai frecvent când ajung la vârsta adultă.
- insuficiență respiratorie datorată problemelor pulmonare.
- Pierderea auzului.
- Modificări ale vederii, care pot fi însoțite de strabism.
- Vărsături.
- Constipație
- Convulsii.
Cum poate fi diagnosticată?
Acest sindrom apare de la concepție, chiar dacă atunci când copilul se naște, simptomele nu sunt clare și sunt mai subtile decat in copilarie tarzie sau la maturitate; mai ales dacă acestea sunt forme mai blânde ale bolii. Din acest motiv, este detectat târziu de mai multe ori.
Cu toate acestea, cele mai frecvente este această condiție și suspectate la scurt timp după naștere de malformații, de obicei, prezent (Steiner, 2015).
Potrivit Organizatiei Nationale pentru Tulburari rare (2016) Diagnosticul se bazează pe examenul fizic si un test de sange pentru a detecta nivelul de colesterol. Este esențial ca copilul să fie evaluat în toate aspectele posibile asociate bolii, cum ar fi ochii, urechile, inima, mușchii scheletici, organele genitale și tulburările gastro-intestinale.
În ceea ce privește testele de sânge, un subiect cu SLO va avea o concentrație mare de 7-dehidrocolesterolului (7-DHC) din sânge (precursor să fie transformat de enzima reductaza 7-dehidrocolesterolului pentru colesterol) și nivelurile de bine Colesterolul scăzut
Acesta poate fi, de asemenea, detectate inainte de nastere prin ultrasunete sau cu ultrasunete tehnica, un dispozitiv care foloseste undele de sunet pentru a examina interiorul uterului gravidă. Prin această tehnică, se pot observa deformările fizice ale acestui sindrom.
Un alt test este amniocenteză, constând dintr-o extragere a unui mic eșantion de lichid amniotic (dond fatul se dezvolta) defect detectarea tip genetic.Aceleași informații pot fi obținute prin eșantionarea villusului corionic (CVS), prin extragerea unei mostre de țesut de la placentă.
Pe de altă parte, testele genetice moleculare pot fi folosite pentru diagnosticul prenatal pentru a observa dacă există mutații în gena DHCR7 și dacă aceasta va prezenta boala sau va fi doar un purtător.
Care este cursul bolii?
Din păcate, majoritatea cazurilor mai severe de SLO mor la scurt timp după naștere. Dacă există dizabilități intelectuale severe, este dificil pentru acești oameni să dezvolte o viață independentă.
Cu toate acestea, dacă se acordă o atenție medicală adecvată și o dietă bună, acești pacienți pot duce o viață normală.
Ce tratamente există?
În prezent, nu există un tratament specific pentru sindromul Smith-Lemli-Opitz. Acest lucru se datorează faptului că originea biochimică a bolii nu este cunoscută astăzi cu certitudine absolută, deoarece colesterolul are mai multe funcții complexe în metabolism.
Tratamentul medical pentru SLO se bazează pe problemele specifice care se regăsesc în copilul afectat și este mai bine să intervină mai devreme.
Poate fi foarte util să primiți suplimente de colesterol sau să creșteți consumul de colesterol prin dietă, pentru a îmbunătăți nivelul de dezvoltare și pentru a diminua fotosensibilitatea. Uneori se combină cu acizi biliari.
Pentru intoleranța la soare, este recomandabil ca acești pacienți să utilizeze ochelari de soare, ochelari de soare și îmbrăcăminte adecvată atunci când merg în exterior.
Sa demonstrat că aprovizionarea cu medicamente, cum ar fi simvastatina, poate reduce severitatea bolii. Deși fenotipul clinic apare în timpul absenței colesterolului în embriogeneză, acesta trebuie administrat la acel moment (Witsch-Baumgartner & Lanthaler, 2015).
Pe de altă parte, poate fi utilizat și un medicament antagonist al precursorului toxic al colesterolului care este în exces (7-dehidrocolesterol) pentru a preveni creșterea acestuia. Suplimentele cu vitamina E pot ajuta.
Alte tipuri de medicamente specifice pot fi utile pentru simptome precum vărsături, reflux gastroesofagian sau constipație.
Chirurgia sau talpa ar putea fi necesare dacă există deformări fizice sau probleme musculare legate de acest sindrom, cum ar fi palate de la nivelul gurii, defecte cardiace, hipotonie musculară sau alterarea genitală.
În concluzie, este necesar să continuăm cercetarea acestui sindrom astfel încât să se dezvolte tratamente mai eficiente și mai specifice.
referințe
- Jiménez Ramírez, A.; Valdivia Alfaro, R .; Hernández González, L .; León Corrales, L .; Machín Valero, Y. și Torrecilla, L. (2001). Smith Lemli Opitz. Prezentarea unui caz cu diagnostic biochimic. Espirituana Medical Gazette, 3 (3).
- Smith Lemli Opitz. (N.d.). Recuperat la 6 iulie 2016 de la Organizația Națională pentru Tulburări Rare (NORD).
- Smith-Lemli-Opitz. (N.d.). Adus la 6 iulie 2016 de la Universitatea din Utah, Științe ale Sănătății.
- Smith-Lemli-Opitz. (N.d.). Adus la 6 iulie 2016 de la Consiliu.
- Sindromul Smith-Lemli-Opitz. (5 iulie 2016). Adus de la Genetics Home Reference.
- Steiner, R. (1 aprilie 2015). Smith-Lemli-Opitz. Obținut de la Medscape.
- Tint, G.S., Irons, M., Elias, E.R., și colab. (1994). Defecțiunea biosintezei colesterolului asociată cu sindromul Smith-Lemli-Opitz. N Engl J Med, 330: 107-113
- Witsch-Baumgartner, M., & Lanthaler, B. (2015). Ziua de naștere a unui sindrom: 50 de ani de la sindromul Smith-Lemli-Opitz. European Journal of Human Genetics, 23 (3), 277-278.