Acțiune:



Sindromul Rokitnasky-Küster-Hauser Simptome, cauze și tratamente
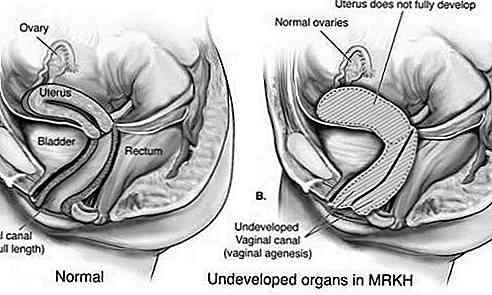
Sindromul Rokitansky-Küster-Hauser (MRKH) este o tulburare care afectează sistemul reproductiv feminin caracterizat prin subdezvoltarea sau absența uterului și a vaginului.
Femeile cu acest sindrom dezvoltă caracteristici sexuale secundare în timpul pubertății - senos, păr pubian - dar nu au un ciclu menstrual (amenoree primară).
Nu începe ciclul menstrual, este adesea semnul inițial al sindromului MRKH. Deși femeile cu această afecțiune nu pot avea sarcini, dacă pot avea copii prin reproducere asistată.
Severitatea sindromului poate varia în funcție de tip. Tipul 1 este caracterizat printr-o absență izolată a două treimi proximale ale vaginului. Tipul II se caracterizează prin alte defecte, cum ar fi anomalii renale (40% din cazuri), anomalii ale scheletului (20-25%), depreciere (10%) de auz, mai rar, defecte cardiace.
Datorită naturii bolii, este posibil să provoace probleme psihologice semnificative, deci este recomandat să cereți ajutor.
Sindromul MRKH are o incidență mondială de 1 din 4500 de nașteri ale femeilor, conform diferitelor studii.
Este, de obicei, diagnosticată mai frecvent la adolescență, când se verifică dacă ciclul menstrual se dezvoltă. Deși femeile nu pot avea copii, ovarele lor sunt normale și funcționale.
Simptomele sindromului Rokitnasky-Küster-Hauser
Simptomele sindromului MRKH variază foarte mult de la o femeie la alta, deci rețineți că femeile afectate pot să nu aibă toate simptomele menționate mai jos.
Sindromul Mayer-Rokitansky-Hauser de tip I
Acest tip este, de asemenea, cunoscut ca Aplasia lui Müller și se caracterizează prin dezvoltarea inadecvată a uterului și a vaginului. În cele mai multe cazuri, uterul și / sau vaginul nu s-au dezvoltat (aplasie); sau există o îngustare a părții superioare a vaginului și a uterului (atrezia). Tuburile uterine pot fi, de asemenea, afectate.
Unele dintre caracteristicile sau simptomele sindromului MRKH sunt:
- Prima menopauză sau absența perioadelor în timpul pubertății. Deși acest simptom este comun în sindromul MRKH, pacientul suferă de pubertate cu telarca și adrenarche normale. Cu toate acestea, nu există menstruație.
Deoarece funcția ovarelor este normală, pacienții se confruntă cu toate schimbările care sunt asociate cu menstruația sau pubertatea.
Organele genitale externe normale.
- Reducerea adâncimii vaginale, de la 2 la 7 cm.
- Caracteristicile sexuale, cum ar fi sanii normali și părul pubian.
-Ovarii functionale cu nivele normale de estrogen.
Modele cromozomiale normale.
- În sindromul MRKH de tip 1, numai vaginul și uterul sunt anormale. In MRKH tip II, defecte in vagin si uter poate fi însoțită și de anomalii în trompa uterină, la fel ca în rinichi sau coloanei vertebrale.
- Deși anomaliile cardiace sunt rare, poate apărea fereastra aortei-pulmonare, defectul septal atrial și stenoza valvei pulmonare
-Pacienții cu sindrom MRKH se plâng, de obicei, de dureri abdominale ciclice datorate detașării endometrului ciclic fără o cale de drenaj de brevete.
-Esteterilitatea: Mulți pacienți solicită adesea îngrijire clinică pentru infertilitate, dar nu pentru amenoreea primară.
- Un alt simptom este dificultatea relațiilor sexuale, deoarece gradul de aplazie vaginală poate varia de la absența completă până la dezvoltarea slabă, care poate provoca dispararenie.
- Dificultăți de urinare, incontinență urinară, infecții urinare recurente.
- Anomalii vertebrale.
- O slingă de țesut palpabil poate fi prezentă la nivelul reflexiei peritoneale.
- Malformații renale.
Simptomele sindromului MRKH de tip II
Insuficiența renală este cea mai comună anomalie asociată cu sindromul MRKH de tip II.
Femeile cu sindrom MRKH tip II poate avea nici un rinichi, malformații ale unuia sau ambilor rinichi (displazia renala), in curs de dezvoltare rinichi (hipoplazie) și / sau plasarea incorectă în interiorul corpului unuia sau a ambilor rinichi (ectopie renală).
Aceste anomalii pot provoca deficit de creștere renale, pietre la rinichi, sensibilitate crescută la infecții ale tractului urinar și acumularea anormală de urină în rinichi din cauza obstrucției.
Multe femei cu sindrom MRKH de tip II pot avea, de asemenea, malformații scheletice, cum ar fi probleme osoase în vertebrelor cervicale și toracice care se pot dezvolta în mod necorespunzător.
pot apărea anormalități ale feței, cum ar fi având o falcă anormal de mici (micrognathia), buza cleft si palatului, iar nedezvoltarea o parte a feței, rezultând asimetrie facială.
Multe femei afectate pot dezvolta, de asemenea, probleme de auz, în principal datorită anomaliilor structurale ale urechii medii.
Când sunt implicate urechile, tulburarea poate fi numită sindromul urechii genitale a urechilor (GRES).
Unele dintre femeile cu sindrom MRKH de tip II au avut anomalii fizice suplimentare care includ defecte ale mâinilor și / sau brațelor.
Aceste anomalii la nivelul extremităților pot include absența unuia sau mai multor degete și degete, duplicat degetul mare și absența osului antebraț lung și subțire (raza absentă). Nu toate simptomele apar la toți pacienții, depinde de localizare și severitate.
cauze
În cele mai multe cazuri, originea sindromului MRKH este necunoscută și apare la femeile fără istoric familial.
Cercetătorii indică factorii genetici și de mediu ca fiind cauzele sindromului, deși nici o genă sau gene asociate cu această afecțiune nu au fost încă determinate.
Din punct de vedere istoric, cercetatorii au sugerat ca sindromul poate aparea ca urmare a unei boli materne sau datorita expunerii fetale la diferite substante nocive, cum ar fi anumite medicamente, abuzul de droguri, alcoolul ...
Cu toate acestea, nu a existat nici un studiu care să confirme această asociere între sindrom și consumul altor medicamente.
În unele familii, sindromul pare să aibă un model dominant de moștenire. Cu toate acestea, modelul de moștenire este dificil de stabilit, deoarece nu toate femeile care suferă de el au aceleași simptome, chiar dacă sunt din aceeași familie.
Potrivit cercetătorilor, este cel mai probabil o combinație de factori genetici și de mediu.
În ceea ce privește anomaliile în procesul de reproducere, acestea se datorează unei dezvoltări incomplete a conductei Mülleriene, însă cauza ei rămâne necunoscută.
Se pare că în ultimii ani sa înregistrat o creștere a dovezilor, deoarece sindromul MKRH este o tulburare genetică. Cresterea studiilor de caz a insemnat ca aceasta idee continua sa fie intarita.
Vorbim despre tulburări genetice atunci când există o combinație de gene pentru o anumită trăsătură. În cazul în care este o tulburare genetică dominantă, așa cum ar fi în acest caz, ar fi produsă de dezvoltarea anormală a unei singure copii a unei gene anormale.
Această gena defectă poate fi moștenită atât de mamă, cât și de tatăl sau se poate datora unei noi mutații în gena însăși.
Probabilitatea transmiterii genei defecte va fi de 50% pentru fiecare sarcină, indiferent de sexul copilului.
Moștenirea poligenă a fost de asemenea propusă ca o cauză a sindromului MRKH.
Până în prezent, s-au găsit șapte ștergeri și o dublare a segmentelor cromozomiale la mai multe persoane afectate de sindromul MRKH. Dar numai una dintre aceste anomalii a fost găsită pe persoană.
În prezent, acești cromozomi au fost identificați acolo unde este posibil ca eliminările segmentate să fie implicate. Aceste cromozomi sunt: cromozomul 1 (1q21.1), 4 (4q34q), 8 (8p23, 1), 10 (10p14-15), 16 (16p11.2), 17 (17q12) și 22 (22q11.21) , iar duplicarea a fost găsită pe cromozomul X (Xpter-p22.32).
Toate aceste informații au determinat cercetătorii să selecteze mai multe gene candidate, printre care: HNF1B, Lhx1, TBX6, ITIH5 și SHOX.
tratament
Obiectivul tratamentului este acela că pacientul are o funcționare sexuală completă și satisfăcătoare.
Deoarece simptomele sunt atât de variabile, este necesară colaborarea unei echipe de specialiști, pentru a asigura o abordare cuprinzătoare a tratamentului.
Ca regulă generală, pacienții cu sindrom MRKH sunt încurajați să caute ajutor psihologic după diagnosticare și înainte de tratament, deoarece acest sindrom poate provoca anxietate sau suferință psihologică. De asemenea, sunt recomandate grupuri de suport.
În ceea ce privește tratamentul aplaziei vaginale este crearea unui vagin nou.
Tratamentul poate fi chirurgical sau nu. Non-chirurgical va fi întotdeauna prima opțiune. Una dintre opțiuni sunt dilatorii vaginali, care servesc la mărirea sau crearea unui vagin.
Cea mai cunoscută metodă este dilatorul Franck, cunoscut și ca dilatatorul perineal, care nu necesită nici un fel de operație chirurgicală.
Fiind auto-administrat pacientul trebuie să fie suficient de motivat să îl folosească. Aceasta durează aproximativ șase săptămâni până la câteva luni.
O altă opțiune poate fi o vaginoplastie, care ar crea un vagin superficial. Dar despre această intervenție chirurgicală nu există prea mult acord asupra tehnicilor de utilizat.
Tehnica Mclndoe este una dintre cele mai utilizate proceduri chirurgicale pentru reconstrucția vaginului. O grefă de piele luată de la fesă sau coapsă este luată și aplicată pe o proteză în formă de penis care este gonflabilă. Aceasta proteza grefa cu mulează tunel vaginal, care se deschide cu un bisturiu și disecat în mod de tăiere și bont, cu grijă să nu deteriora vegija, rect sau peritoneu.
Este lăsat șapte până la zece zile și când este schimbat, este sub anestezie. Ulterior, pacientul va folosi alte proteze de dilatare. La trei luni pacientul poate face sex.
Problema cu această tehnică este că, în plus față de părăsirea cicatricilor, este necesară dilatarea pe termen lung.
O altă tehnică utilizată pentru sindromul MRKH este neo-vaginul intestinal.
Această tehnică utilizează un segment izolat al intestinelor. Se compune din secționarea unui fragment al colonului sigmoid printr-o incizie abdominală și transferarea acestuia în zona în care a fost creat spațiul neovaginal. Această tehnică este destul de complexă și poate dura până la 8 ore.
Avantajul acestei tehnici este acela că lasă un vagin spațios de o lungime adecvată și, de asemenea, că este auto-lubrifiant. În ceea ce privește dezavantajele, primul ar fi o intervenție chirurgicală destul de complexat și infrabdominal, lăsând secreții mucoase exces fragmentului intestinal, prolapsul vaginal mucoide și inflamația mucoasei la neovagina.
În cele din urmă, alte tehnici sunt Tehnica Vecchieti.
Această tehnică exercită o presiune continuă progresivă de către o măslină acrilică prin puterea spațiului neovaginal și peretele abdominal. Apoi, un dispozitiv de tracțiune este plasat în cavitatea peritoneală și îndepărtează treptat bolta vaginală. Această tehnică se face acum prin laparoscopie.
referințe
- https://visualsonline.cancer.gov/
- https://ghr.nlm.nih.gov/condition/mayer-rokitansky-kuster-hauser-syndrome
- http://emedicine.medscape.com/article/953492-overview
- http://www.news-medical.net/health/Mayer-Rokitansky-Kuster-Hauser-(MRKH)-Syndrome-Symptoms-(Spanish).aspx
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=3109
- https://rarediseases.org/rare-diseases/mayer-rokitansky-kuster-hauser-syndrome/
- http://www.biomedcentral.com/content/pdf/1750-1172-2-13.pdf
- Imaginea sursei: http://www.mrkhnorge.org/mrkh/?lang=en